肠道微生物由细菌、古菌、真菌和病毒等组成。其中,肠道病毒是其它三类微生物的重要调控者,且可作为抗生素的替代品用于致病菌感染的治疗,具有重要理论研究意义和应用价值。虽然在人类肠道病毒基因组的识别上已取得了显著成就,但与原核微生物相比,对肠道病毒组的表征明显不足。尤其是,生物学与技术方面的挑战导致了肠道病毒多样性被严重低估。目前,宏病毒组测序主要采用两种方法:全微生物群落测序(宏基因组学)和病毒样颗粒富集测序。全微生物群落测序利用生物信息学工具,直接从宏基因组数据集中鉴定病毒基因组,这种方法倾向于获得高丰度的病毒,而忽略那些地丰度和尚未被充分描述的病毒。相反,病毒样颗粒富集测序通过富集病毒样颗粒来有效减少宿主及细菌基因组污染,这有助于发现低丰度和新颖病毒序列。然而,该方法通常依赖于全基因组扩增(WGA)来获取足够的DNA进行测序,这一过程存在明显缺陷,包括基因组覆盖的不均衡性、嵌合序列的形成及扩增偏好性。
第三代测序技术因能够生成更长的读长而被广泛采用。将第三代测序技术应用于肠道病毒样颗粒富集测序,能显著增强对低丰度及新颖病毒序列的发现,从而深化我们对肠道病毒群落多样性的认识,并拓宽我们对人类肠道微生物生态学中病毒暗物质的了解。
2024年1月19日,华中科技大学生命学院陈卫华教授团队在top期刊《Advanced Science》在线发表题为“Efficient Recovery of Complete Gut Viral Genomes by Combined Short- and Long-Read Sequencing”的学术论文。

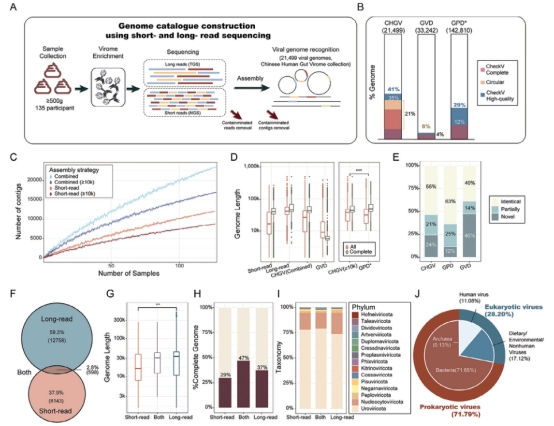
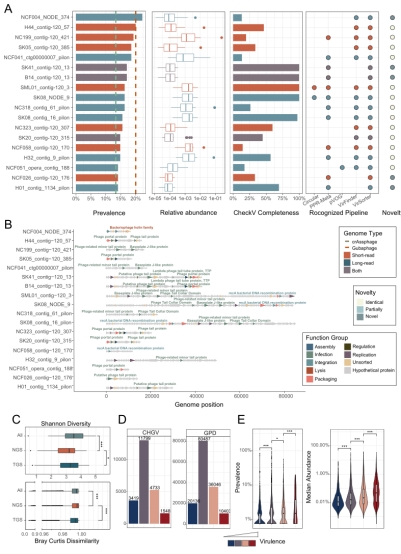
该研究开发了一种肠道病毒组检测流程,包括病毒样颗粒富集和二三代测序;并将之应用于135个粪便样本,构建了中国肠道病毒组目录(CHGV)。该数据集中包含21,499个非冗余病毒基因组序列。相较于现有公共数据集,CHGV中的病毒基因组序列平均长度更长,且超过1/3为完整病毒基因组,该比例为GVD(Gut Virome Database)数据集的9倍。此外,研究发现了6,962个新的病毒基因组,这些新病毒基因组与现有公共病毒数据集没有同源性。在CHGV中,还发现了几十种在肠道中高流行度的噬菌体,其流行率超过了在人类肠道中最流行的crAssphage (~20%)和Gubaphage。高度的流行率暗示其可能塑造肠道微生态群落中有着重要作用。此外研究还发现比crAssphage和Gubaphage具有更高多样性的病毒簇。总体而言,这些结果强调了二三代测序联用在揭示肠道病毒基因组多样性方面的重要性,并支持了肠道病毒研究和应用的进一步发展。
华中科技大学生命学院陈卫华,刘智教授和复旦大学赵兴明教授为共同通讯作者。华中科技大学生命学院博士生陈景超,孙楚晴,金梦露为第一作者。华中科技大学生命学院为第一完成单位。
近年来,华中科技大学生命科学学院陈卫华教授团队致力于深入研究肠道病毒组、肠道噬菌体的基因工程以及肠道微生物群的调控机制,以期为理解肠道病毒群落组成与健康关系提供科学依据,为通过噬菌体精准调控肠道菌群、服务人体健康提供工具。近5年,该团队的研究成果已在包括《Advanced Science》、《Gut》、《Microbiome》、《Genome Biology》、《Nature Communications》、《Nucleic Acids Research》、《Cell Host & Microbe》、《Cell Metabolism》和《Gut Microbes》等国际知名学术期刊上发表。
该研究得到了国家自然科学基金、国家重点研发计划、中瑞(NNSF-VR)合作研究项目基金以及上海市科技重大项目的支持。
论文链接:https://onlinelibrary.wiley.com/doi/10.1002/advs.202305818
