蛋白质是一切生命的物质基础,是生命活动的直接执行者。解析蛋白质结构和其对应的功能是生命科学领域的一个经久不衰的课题。虽然Alphafold等从头预测蛋白3D结构方法的提出,能够大幅提高蛋白质3D结构预测的准确性,但是仍然有很多蛋白缺乏足够的同源序列,无法对其进行结构预测,导致当前预测效率和完整性尚不理想。目前生物学家逐渐认识到微生物组大数据储藏着大量未知结构蛋白的同源序列,并且最近许多工作已经成功利用微生物组大数据补充未知结构蛋白的同源序列,从而预测出大量之前未知结构蛋白的可靠三维结构,极大推动了蛋白结构预测领域的发展。但是,随着微生物组大数据呈指数型增长,盲目的全盘使用微生物组数据使得搜索同源序列以及结构预测变得越来越低效,而且整个过程不具有良好的生物解释度。
日前,华中科技大学生命科学与技术学院宁康教授团队联合美国密歇根大学计算医学与生物信息系张阳教授团队,在利用微生物组大数据靶向高效辅助预测蛋白质三维结构领域取得突破。该联合团队研究成果《Decoding the link of microbiome niches with homologous sequences enables accurately targeted protein structure prediction》在国际知名学术杂志美国国家科学院院刊(Proceedings of the National Academy of Sciences of the United States of America,PNAS) 于2021年12月7日在线发表。宁康教授、张阳教授为并列通讯作者,博士生杨朋硕和博士后郑伟为并列第一作者。

该研究通过解析不同生态位下的微生物组与蛋白结构的关系,在世界上首次在宏观层面发现了来源于微生物组的同源序列在不同生态位中的不均衡分布和富集特征;进而利用富集特征构建机器学习模型用于预测未知结构蛋白的同源序列的来源微生物组的生态位,高效补充同源序列。通过收集42.5亿的微生物组蛋白(来自地球上最主要的四个生态位:肠道,湖水,土壤,发酵罐),该团队预测出了1,044个未知结构蛋白的可靠结构(占当前Pfam蛋白质家族数据中未知结构蛋白总数的约1/8)。通过解析微生物组生态位与预测出的1,044个蛋白结构的关联后,发现不同生态位的微生物组数据对不同蛋白结构的预测能力存在差异。而这一预测能力的差异通过对微生物数据补充同源序列的边际效应和微生物数据的利用效率的评估得到进一步的定量描述。于是,该研究团队基于靶向性人工智能建模,构建预测器(MetaSource),来预测未知结构蛋白在哪个生态位的微生物组存在最多同源序列。经过已知结构蛋白的验证,MetaSource能够准确指导其同源序列的补充。对比使用所有42.5亿蛋白构建的模型,使用MetaSource 能够显著降低比对时间(MetaSource:每个蛋白比对0.74TB微生物蛋白序列; 所有数据: 每个蛋白比对2.40TB微生物蛋白序列),同时MetaSource得到的模型具有更高的接触预测准确性(contact accuracy:MetaSource:0.512;所有数据:0.496)和模型预测准确性(TM-score: MetaSource:0.625;所有数据:0.609)。
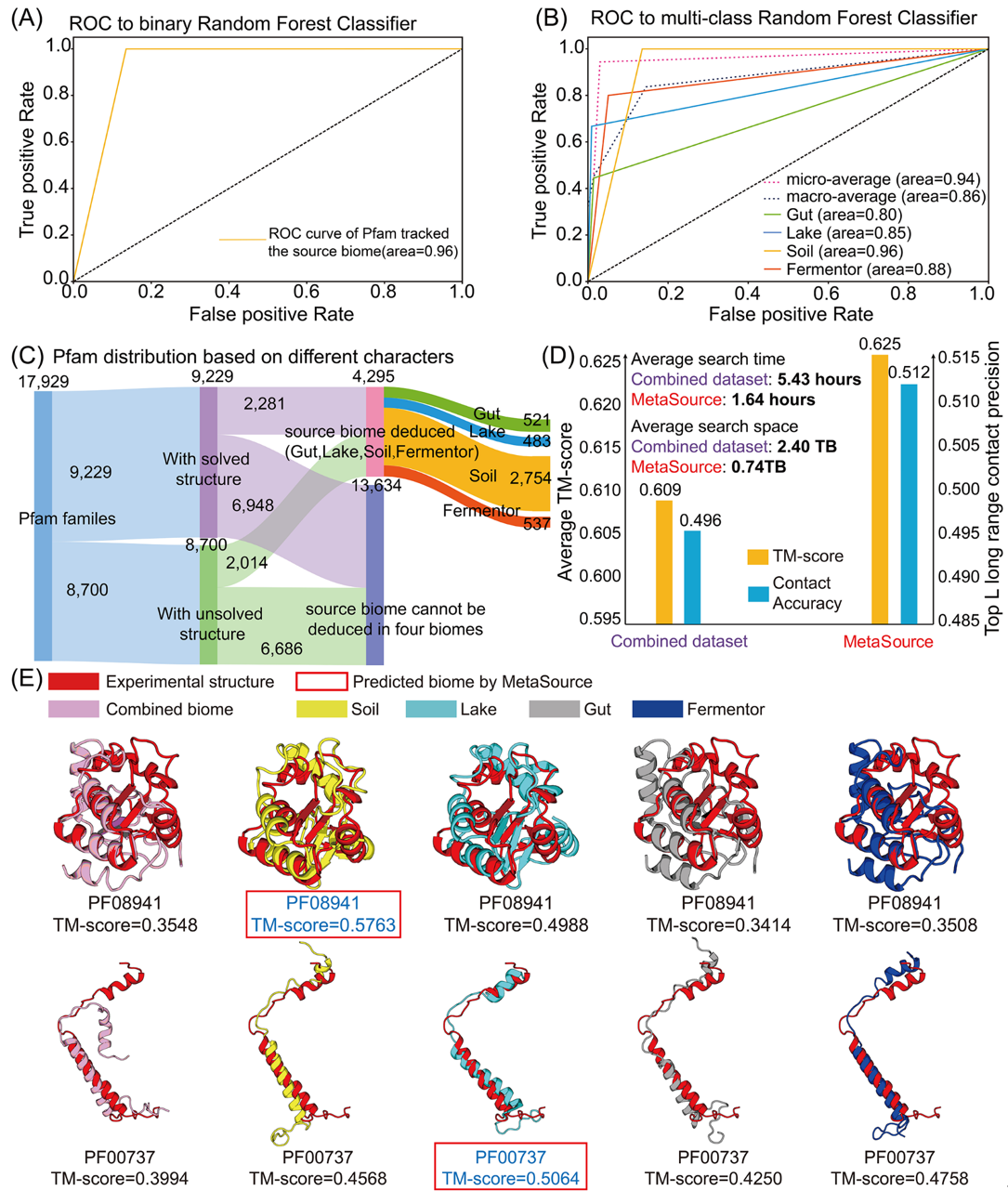
构建的MetaSource 模型可以为 Pfam 家族预测其同源序列的来源生态位。(A) 二元分类 MetaSource 模型的准确性分析。构建此模型是为了检测查询的Pfam 家族的来源生态位是否是四个生态位之一。 (B) 多分类 MetaSource 模型的 准确性分析。该模型旨在预测 Pfam 家族的来源生态位。为了评估整体预测精度,应用了微观平均值(通过汇总所有类别的贡献来计算平均指标)和宏观平均值(由每个类别的指标独立计算并取平均值)。 (C) 基于 MetaSource 模型预测结果的所有 Pfam 家族的 Pfam 分类结果。 (D) 组合和 MetaSource 预测的生物群落数据集的平均 TM-score、top-L contacts的准确性和平均 MSA 搜索时间。 (E) PF08941 和 PF00737作为示例来说明使用来自不同生物群落的 MSA 对 Pfam建模精确度有差异。红框是MetaSource预测出的存在最多同源序列的环境。
该研究是系列研究“利用宏基因组数据和人工智能技术辅助蛋白质结构预测”的第二期工作。在第一个研究中,该合作团队利用海洋微生物组大数据辅助预测蛋白结构,发现预测出可靠结构的蛋白结构参与海洋微生物组中重要的光合作用通路,初步揭示了微生物组与构建的蛋白结构之间的关系,一期研究于2019年11月发表在生物学领域权威期刊Genome Biology。
而作为二期工作,该研究在更加宏观层面验证了这一重要的微生物组-序列-蛋白结构之间的关联性,并且构建了基于机器学习的靶向方法来更高效地利用微生物组辅助预测蛋白质结构和功能。一方面,该工作可以作为利用宏基因组序列辅助预测蛋白结构并解析其潜在功能的”蓝皮书”,为进一步完善蛋白质结构预测方法指出了方向。另一方面,该工作首次发现了来源于微生物组的同源序列在不同生态位中的不均衡分布和富集特征,为更深入的理解功能基因的适应性进化提供了新的视角。
本项目依托于华中科技大学生命科学与技术学院、人工智能生物学中心(筹)等单位开展,受到了科技部重大研究计划、国家自然科学基金委项目等的支持。
参考文献:
Pengshuo Yang, Wei Zheng, Kang Ning* , Yang Zhang*. Decoding the link of microbiome niches with homologous sequences enables accurately targeted protein structure prediction. Proceedings of the National Academy of Sciences of the United States of America (PNAS), 2021, 118 (49) e2110828118.
Yan Wang, Qiang Shi, Pengshuo Yang, Chengxin Zhang, Golam Mortuza, Zhidong Xue*, Kang Ning*, Yang Zhang*. Fueling ab initio folding with marine microbiome enables structure and function predictions of new protein families.Genome Biology , 2019,Nov 1;20(1):229.
第一作者在线讲解链接为http://microbioinformatics.org/pnas.mp4(为获得最好的观看效果,请用电脑浏览器打开,例如chrome或Microsoft edge)。


