近日,由来自德国埃尔朗根-纽伦堡大学、加州大学旧金山分校以及香港中文大学(深圳)科比尔卡药物创新研究院等高校的研究人员通过大规模虚拟筛选、药理学以及药代动力学实验,发现了靶向a2A肾上腺素受体的新型小分子激动剂。对比传统的a2A药物,这些新型药物在镇痛剂量下不会引起镇静且可口服,从而大大增加了其在疼痛治疗中得到更广泛使用的可能性。这项研究也证明,a2A肾上腺素受体引起的镇痛和镇静效果,可以被某些药物区分开来。可以说,此次关于药物前体的开发,为非阿片类药物疼痛疗法带来了新的曙光。未来的新型小分子化合物、或后续进一步优化的小分子药物,有一定的潜力成为新型止痛药从而被直接应用于临床。
华中科技大学生命科学与技术学院2010级生物技术专业本科生徐俊,是此项研究的核心成员,并以共同一作的身份将相关研究成果发表在Science上。2014年徐俊本科毕业后被保送至清华大学攻读生物学博士学位。期间,他有幸加入到2012年诺贝尔化学奖得主、斯坦福大学医学院分子与细胞生理系教授布莱恩科比尔卡(Brian Kobilka)在清华的实验室开展GPCR的动态结构研究。因在博士期间的优异表现,毕业后受到Brian Kobilka教授邀请,前往其在斯坦福大学医学院的实验室进行博后研究,继续从事有关GPCR结构、动态以及药物研发的工作。

一、对非阿片类止痛药的开发,具有潜在的临床意义。
据介绍,G蛋白偶联受体(GPCR,GProtein-CoupledReceptors),是人体内最大的细胞表面受体家族,它们通过感知细胞外的多种信号分子,包括各种神经递质、多肽、脂质,甚至光和气味,将细胞外信号传递至胞内,从而调控细胞特定的生理变化。该家族受体广泛分布于人体的各个部位,几乎参与人体所有的生理和病理过程,因此一直以来是非常重要的药物靶标。
据统计,市场上有30-40%的药物是靶向GPCR的。GPCR的信号传导,需要细胞内的信号蛋白分子介导。G蛋白以及阻遏蛋白arrestin,是两种主要的可以结合GPCR、并能将信号传递至细胞下游的蛋白分子。近年来的研究发现,这两条信号通路的激活往往会导致不同的生理反应。对于药物分子来说,它能通过激活GPCR、产生多样化的生理反应,从而发挥药效。但在同时,也会产生各种副作用。因此,学界尝试开发一种具有功能选择性、或者偏向性的配体药物。受体在被这种药物激活之后,可以选择性地结合下游某种特定的信号蛋白,比如只激活G蛋白而不激活阻遏蛋白,从而只产生发挥药效的生理反应,但却可以避免其他信号通路产生的副作用。
以阿片受体为例,当其激活G蛋白信号通路的时候,可以产生很好的镇痛效果。但是,阻遏蛋白信号通路却能产生呼吸抑制、便秘等副作用。同时,阿片类药物还具有成瘾性等副作用。
疼痛是所有疾病中最为常见的症状之一。研究表明,疼痛已成为继心脑血管疾病、肿瘤之后的第三大健康问题,严重影响着人们的健康和生活质量。阿片类药物,是最有效也最常用的治疗疼痛的药物。然而,由于其副作用过于明显,过去十年来阿片类药物滥用所导致的死亡人数增加了近四倍。因此,科学家们一直在努力开发更加安全有效的阿片类药物,期望在保持其镇痛效果的同时,可以降低各种副作用。
偏向性配体药物的开发,是其中的一个重要的方向。与此同时,学界也在尝试针对新的镇痛靶点,开发更加安全有效的非阿片类药物。作为GPCR的其中一类,a2A肾上腺素受体可以感知人体中重要神经递质肾上腺素和去甲肾上腺素,它们广泛分布在人体的外周器官和中枢神经系统中,可以调节一系列重要的生理功能,包括血压、血糖、心率等。同时,其在中枢神经系统中的激活,可以产生缓释疼痛的效果,是潜在的镇痛靶点。一些传统的a2A激动剂药物比如右旋咪唑具有一定的镇痛效果。
但是,其镇静作用过强、以及不能口服的缺陷,限制了其作为临床镇痛药物的使用。早期研究发现,敲除阻遏蛋白arrestin可以减弱a2A药物的镇静作用。因此,开发出只激活G蛋白的偏向性配体药物,或许可以保留其镇痛作用,并能避免或降低镇静效果。
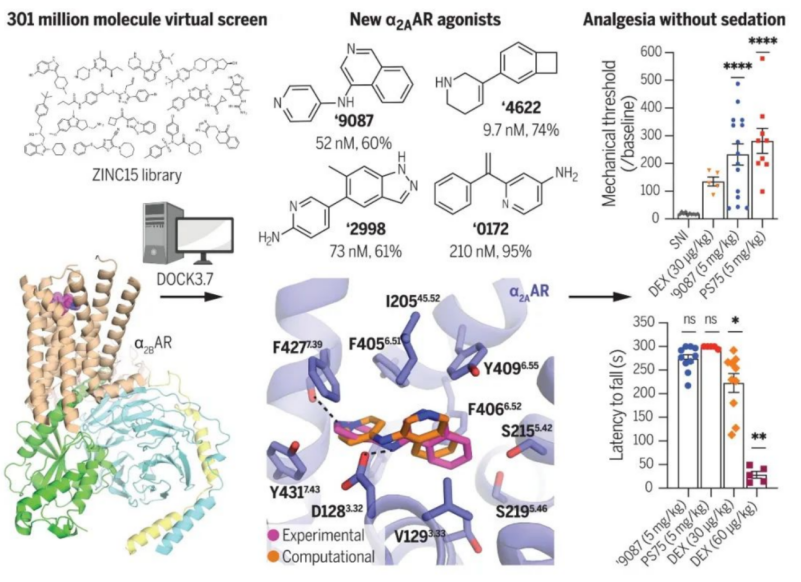
基于此,徐俊和所在团队开展了这项靶向a2A肾上腺素受体药物开发的研究。为了寻找这种潜在的、具有特殊生物学功能的新型激动剂,他们利用分子对接的方法,针对与a2A同家族的a2B肾上腺素受体,进行了大规模的计算机虚拟筛选实验。
此前,清华大学医学院团队解析的a2B肾上腺素受体与右旋咪唑的原子分辨率三维结构,清晰的阐释了药物分子右旋咪唑的结合口袋、以及与受体的结合模式。[1]。清华团队解析出来的结构,也为此次徐俊及其合作者的虚拟筛选实验提供了精细的模版。他和所在团队从近3亿个小分子虚拟文库中,筛选出了48评分较高的小分子。幸运的是,初期的药理学实验显示,其中有63% 的小分子与a2B具有较强的结合能力,35%的小分子则与a2A具有较强的结合能力。
令课题组惊讶的是,其中结合能力较强的几个小分子激动剂均,显示出一定的功能选择性、即只激活G蛋白,但是募集阻遏蛋白arrestin的活性却很弱。进一步地,他们利用冷冻电镜技术解析了其中2个功能选择性的小分子与a2A的精细三维结构,从而揭示了其作用模式,并以此为模板进行了进一步的优化,得到了结合能力更强的、且功能选择性更好的类似物。后续的动物实验显示,在神经性和炎症性疼痛的小鼠模型中,这些新型小分子激动剂均展现出良好的镇痛效果。并且,相比于右旋咪唑,患者可以口服且不会引起镇静作用。
近日,相关论文以《基于结构开发靶向α2A-肾上腺素能受体的非阿片类镇痛药》(Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor)为题发表在Science上[2]。

徐俊、美国加州大学旧金山分校博士生伊丽莎·芬克(Elissa Fink)、德国埃尔朗根-纽伦堡大学博士哈拉尔德·哈布纳(Harald Hubner)等担任共同一作。德国埃尔朗根-纽伦堡大学化学与药学系彼得·格梅纳(Peter Gmeiner)教授、美国加州大学旧金山分校药学院布莱恩·肖伊切特(Brian Shoichet)教授、香港中文大学(深圳)医学院/科比尔卡创新药物开发研究院杜洋教授等担任共同通讯作者。
评审专家表示,这项研究运用多个不同的跨学科方法,针对a2A肾上腺素受体展开了药物筛选、药理学以及结构生物学等研究,这些实验数据令人印象深刻,新型小分子药物的发现和前期的实验结果非常新颖,对非阿片类止痛药的开发具有潜在的临床意义。并评价称,这是一项结合了大规模筛选、药物化学、结构生物学、体外和体内药理学的综合性研究。描述了在小鼠疼痛模型中鉴定出具有体内止痛功效的a2A受体激动剂。论文数据揭示,新型化合物具有将镇痛作用和镇静效果分离的潜力。
二、3 所学校、1 个研究
据介绍,该研究经历了小分子筛选及药理学研究、结构生物学研究及小分子优化、体内动物实验等阶段。因为疫情的原因,徐俊在三所不同学校,参与并开展了三个不同阶段的研究。
徐俊在清华大学医学院读博期间,就开始和实验室的师兄袁道鹏博士开展Alpha 2受体、及其下游信号蛋白复合物的结构研究,以更好地理解其信号传导机制、并开展药物研发工作。他们同时研究了Alpha 2 A和Alpha 2 B两个亚型。其中,袁道鹏博士与合作者首先得到了Alpha 2 B与右旋咪唑的高分辨冷冻电镜结构。刚拿到结构模型的时候,徐俊的导师 Brian Kobilka 教授,建议利用该结构进行计算虚拟筛选。对于该结构来说,它是当时冷冻电镜解析的最高分辨率的GPCR、与下游G蛋白的复合物结构。因此在论文发表之前,他们便将结构模型发送给了加利福尼亚大学旧金山分校教授Brian Shoichet 实验室进行分子对接实验。
与此同时,徐俊这边继续进行Alpha 2 A有关的结构研究。2019 年底,课题组完成了虚拟筛选,并由德国埃尔朗根-纽伦堡大学教授Peter Gmeiner 课题组完成了前期的药理学实验。结果发现,在筛选的小分子中,其中多个具备非常好的体外活性和功能选择性。
2020年初,正值新冠疫情爆发,徐俊也因为刚刚毕业而无法返回清华继续开展实验,同时也无法前往美国开展博后研究 。这时,他的导师 Brian Kobilka 教授建议他去香港中文大学(深圳)科比尔卡药物创新研究所,从事一段过渡性研究。“此时,我们实验室的前同事杜洋博士刚刚在港中深建立起自己的GPCR结构药理学实验室,这为该课题的后续研究打下了坚实的基础。”徐俊表示。
在此期间,他们不仅解析了Alpha 2 A受体、与其内源性配体去甲肾上腺素、以及右旋咪唑等传统 Alpha 2激动剂的高分辨结构,也针对刚刚筛选出来的新型小分子化合物,完成了结构解析工作,阐释了其与受体的识别机制,并基于新的结构信息做了进一步的小分子优化工作。“在完成结构生物学研究和优化之后,这时主要由加州大学旧金山分校的Allan Basbaum课题组开展动物实验,并对小分子在体内的活性进行研究。”徐俊表示。
而在此时,徐俊也已经来到斯坦福大学医学院开始博士后研究。在那里,他和大家一起完成了数据分析、论文撰写、投稿等。在后续工作上,一方面针对已有的小分子,合作团队正在进一步开展优化、以及后续的临床实验,以期能够推向临床应用,针对这些小分子也已经申请美国专利。
另一方面,尽管已有的冷冻电镜结构提供了初步的结构基础。但是,对于新型激动剂功能选择性(偏向性)的分子机制,这些静态的结构还解释得不够清楚。除此之外,目前的这些激动剂都不具备亚型选择性,即它们可以同时激活Alpha 2 A、2B、2C受体,这可能会导致一些副作用。因此,开发具备亚型选择的小分子,也是他和所在团队正在努力的方向。
目前,徐俊正利用各种生物物理手段,比如液体核磁、单分子荧光能量共转移、电子双共振等,继续研究这些小分子对受体、以及下游复合物结构的动态调控,以期更好地了解其作用机制,从而为继续优化小分子提供理论指导。
参考资料:
1.D. Yuan, Z. Liu, J. Kaindl, S. Maeda, J. Zhao, X. Sun, J. Xu, P. Gmeiner, H. Wang, B. K. Kobilka. Activation of the α2B adrenergic receptor by the sedative sympatholytic dexmedetomidine. Nature Chemical Biology. 2020 March 09.
2.Fink, E. A., Xu, J., Hübner, H., Braz, J. M., Seemann, P., Avet, C., ... & Gmeiner, P. (2022). Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor. Science, 377(6614), eabn7065.
3. J. Xu, S. Cao, H. Hübner, D. Weikert, G. Chen, Q. Lu, D. Yuan, P. Gmeiner, Z. Liu, & Y. Du. Structural insights into ligand recognition, activation and signaling of the α2A adrenergic receptor. Science Advances. 2022, March
以上内容来源于公众号“DeepTech深科技”推送《新型非阿片类镇痛药诞生,科学家发现靶向肾上腺素受体的新型小分子激动剂,可以口服且无镇静副作用》。
